Introduction
Sickle cell disease (SCD) is a highly variable disorder. Some of the clinical features in patient populations across India, Nepal and the subcontinent might differ from those reported in Sub-Saharan Africa and North America or the UK. Hence, medical care and support need to address these regional differences. Many grassroots-level health practitioners are providing care to sickle cell patients despite the numerous challenges they face, especially in remote, rural and resource constrained settings across South Asia. Medical textbooks, national/state guidelines and protocols often assume ideal healthcare settings and recommend interventions that may be unfeasible and, at times, ignore the resource limitations of health facilities in rural areas. This toolkit has been prepared specifically keeping in view such empirical factors, with the intention of sharing ‘safe practice’ options that can be adapted to your local context. We have collated the information from national and state level policy guidelines, relevant publications as well as discussions over several years between healthcare practitioners, community workers, patients and parents on how to improve long term care.
The practical difficulties and the need for such a guide was first discussed at a two-day long capacity building workshop for community-based organisations and clinicians, “Building Bridges”, held at the Government Medical College Hospital, Nagpur, in November 2019. Despite the National Guidelines link (https://main.mohfw.gov.in/sites/default/files/Final) issued in 2017, different treatment centres or practitioners seemed to follow different treatment protocols. The need for better information and dissemination of guidelines/protocols has also been reported in research (Raman et al., 2021). The background and contents of the toolkit (what to do and what not to do) were discussed at a recent two day training event, “Minimally safe practice toolkit for sickle cell disease” held on the 18th and 19th of March 2022. The workshop was hosted by ASHWINI, Gudalur Adivasi Hospital, in Gudalur and facilitated by a core group of members (SC, NK, DJ, SM and PC). We hope that you will find it useful and would like to get your feedback in a few months’ time.
The toolkit aims to achieve two main objectives:
- First, is to provide an easy summary of the rationale for standard/basic treatment protocols that we know can be put into practice in most district hospitals, primary and community health centres, with limited resources.
- Second, to highlight safe clinical practices that are known to improve outcomes through routine care for sickle cell patients, as evidenced from several centres across India (such as, Government Medical College Hospital, Nagpur; Sewa Rural, Jhagadia; ASHWINI, Gudalur; Jana Swasthya Sahayog , Chhattisgarh; Mission Hospital, Bissamcuttack, and Global Hospital, Mount Abu).
Diagnosis and Screening
There are different testing strategies currently utilised in India with respect to diagnosing sickle cell patients. Further, targeted screening of populations can be conducted to identify potential carriers and patients within community settings. The scope and scale of screening also determines the type of testing that is performed. It is important to note that not all tests are confirmatory. In populations where SCD exists alongside other blood disorders such as beta thalassemia, it is important to use tests that provide clear results such as high performance liquid chromatography (HPLC). Furthermore, screening of children should be prioritised since early detection and treatment is fundamental to improving long term outcomes of care.
Consent before testing should be acquired and information regarding results needs to be clearly communicated. It is also important to have a systematic testing protocol and records to prevent redundant or unnecessary testing. Many communities have been tested multiple times, using different methods for the purposes of scientific research. At times results are not made available to the individuals or families or disclosed in public settings. Such practices erode trust in the system and violate basic principles of ethical conduct. Once a confirmatory test has been obtained, results should be kept safe and available for future use by healthcare professionals and patients. In order to do so the following points should be kept in mind
- SCD patients should always be informed about their results and special care should be taken while communicating to children and their parents.
- Additionally, counselling is always required, including information on risks associated with SCD, where and how to seek necessary treatment and support
- A document, such as a booklet, should be used to make a note of diagnosis details and copies should be kept by the health facility as well as the patient.
When SCD cases are recorded, a unique identification code, usually in an alpha-numeric system, should be assigned to each patient. Often, many facilities use pre-existing unique identifiers assigned by the government such as the Aadhar card. This number can then be used to keep a track of other services that patients receive in the course of their treatment as well. However, it is important to remember that many patients may not have an Aadhar/ identity card and an optional method for record keeping needs to be in place.
Preferably, diagnosis should be provided free of cost, to ensure access to proper medical services. In many settings, tests are subsidised by regional governments or reimbursed by hospitals. This enables a larger number to seek health services. The type of test to be offered depends on the size, scale, and finances available within a medical institution/setting. The table below can be used to determine what approach of testing to take based on such constraints.
| Table 1: Types of tests used to detect SCD status | |||
| Type of test | Type of sample | Facility | Remarks |
| HPLC or Hb Electrophores is | Venous blood sampling Some HPLC machines can detect with heel-prick samples Some machines also take cord blood samples | District hospitals Medical colleges Higher medical institutions | Confirmatory test HPLC is particularly useful for identifying heterozygous sickle cell and beta thalassemia. Requires budgeting for operational costs and maintenance Can be expensive for patients without support. Machines for newborn screening may be calibrated differently. |
| Solubility Test | finger prick sample | Rural health posts Field-based organisations | Non-confirmatory test. Can be used for large population screening. Positive sample means that further testing is required. |
| HPLC/POCT Heel prick/cord blood | Newborn screening for early diagnosis and early treatment | At birth or before discharge, if missed, at first contact with the health visitor. The sample should be dispatched to the newborn screening laboratory. Counselling of parents at diagnosis and from time to time later. | |
| Solubility test / POCT | finger prick sample/venous sample | Antenatal screening (preferably before 12 weeks) | If the woman is positive, solubility test to be offered to the husband. If both are positive, counsel, advise and if willing, refer for prenatal diagnosis. |
Newborn Screening
The aim of new-born screening is to improve outcomes through early treatment and follow up with early administration of
- prophylactic penicillin which markedly reduces the incidence of pneumococcal sepsis in children and
- pneumococcal vaccines which can increase immunity to pneumococcal infections
It allows better follow up and early detection of complications for the patient. In addition, parents can benefit from anticipatory counselling regarding cascade testing within the extended family, prenatal diagnosis and prevention of future births, if so acceptable.
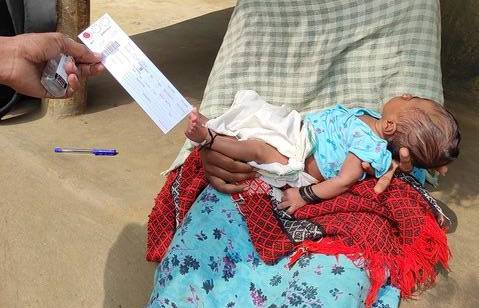
Image 1: Taking a heel prick sample in the community (photo credit, ASHWINI)
Prenatal testing
These tests identify the genotype of the foetus of the target couple/mother by CVS (at 9-12 weeks) and amniocentesis (at 12-18 weeks). Following the test appropriate counselling should be offered to the couple (explained later).
Routine Care of SCD patients (new-born, children and adults)
SCD is a variable condition in terms of frequency and severity of symptoms and complications suffered by patients over a period of time. Often patients might be relatively well for months/years and not attend a medical facility unless/until there is a medical emergency. As with many chronic conditions, they will try and manage their symptoms at home, following different methods of treatment/ healing. It is very important to set up a follow-up protocol to observe them and manage the complications, as summarised below.
Vaso - occlusive crisis and pain management in SCD (children and adults)
Pain, especially, of joints, limbs and abdomen, is a characteristic symptom of SCD (vaso-occlusive crisis or VOC). These episodes, also called sickling or crisis events in literature, can be debilitating and unpredictable in frequency and severity in the same patient over time or across a patient population. Patients and family carers have to be empowered to deal with intermittent as well as recurrent episodes of pain.
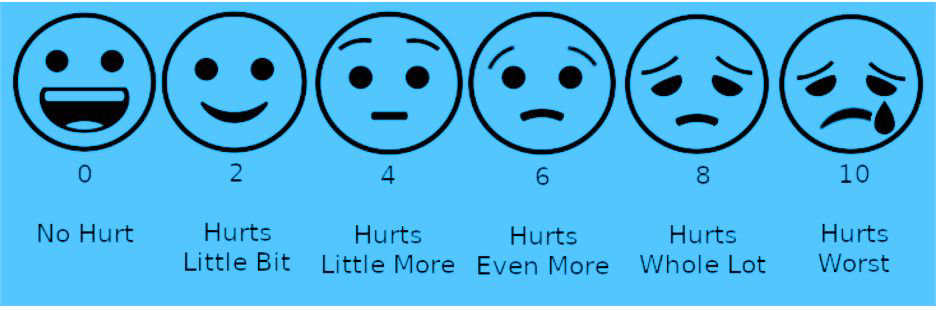
Patients require constant reassurance and comfort, rest for the affected area, and a gentle massage to alleviate pain. Hydration is important and a majority of pain episodes can be managed at home or a primary health centre. It is important to assess the degree of pain and to keep track of how each patient experiences it. Trust your patients when they describe the severity of their pain and use of a pain scale (below) might help them to identify and describe the current pain episode.
- For mild pain: Paracetamol 10 to -15mg /kg every 6 hours
- For moderate pain: Paracetamol every 6 hours with Diclofenac 1mg/kg every 8 hours after food
- For severe pain: Paracetamol 10-15mg /kg AND Diclofenac 1mg /kg and Tramadol 50 to 100 mg orally every 4 to 6 hours for adults above the age of 17. If not responding to the above, patient should go to a hospital
Hospital care for Severe SCD Pain
Each patient has a threshold of pain and their report of pain should be treated as the gold standard. Pain in a crisis is usually quite severe and requires immediate treatment. Do not wait for lab results before starting pain medications; there are no lab values that confirm or rule-out a sickle cell pain crisis. First dose of an analgesic must be given within 30 minutes of arrival of the patient at the health facility. Patient’s requests for specific pain medicines or doses are most commonly due to past experience and not inappropriate drug-seeking behaviour. Pain scales can be useful for assessment (see Table 2 below). Build trust by believing the patient is in pain and show them you are there to help. Empathetic body language is essential (eye contact, facial expression, gestures). Be patient when asking questions; it is often difficult to speak when in severe pain. Pain can make anyone irritable, impatient, or upset.
Severe Pain Management in Adults(>18years)
For severe pain unrelieved by above oral medicines give
- Diclofenac injection 0.5 mg/Kg IM or IV
- Diclofenac injection 0.5 mg/Kg IM or IV and Tramadol hydrochloride injection (1 mg/kg) IV in a total volume of 4 mL (Cochrane Database Syst Rev. 2015)
Table 2. using a simple pain scale to assess the severity of pain
Pain Management in Children (2-18 years)
For severe pain unrelieved by above oral medicines give:
- Diclofenac suppository 0.5 mg/Kg
- IV Paracetamol 5-7mg/kg as a drip over a period of 1 hr.
- Diclofenac injection 0.5 mg/Kg IM or IV if suppository not available AND Tramadol hydrochloride injection (1 mg/kg) IVin a total volume of 4 mL (Erhan et al 2007)
- Diclofenac injection 0.5 mg/Kg IM or IV AND tramadol hydrochloride injection (1 mg/kg) IV, in a total volume of 4 mL.
- Diclofenac is an effective and safe analgesic for acute pain in children.
- The optimum dose of diclofenac for acute pain in children still needs to be ascertained, this review found a range in the single doses being used from 0.5 to 2. 5 mg/kg).
Acute Chest Syndrome
Acute chest syndrome (ACS) is one of the commonest causes of death in SCD among adults. It presents with fever, cough and breathlessness, with a new infiltrate on chest X ray, and falling oxygen saturation.
If suspecting ACS, start oxygen immediately administer with IV ceftrioxone and oral azithromycin, and refer the patient to a centre with an ICU for possible ventilation and exchange transfusion. If transfer is not possible, start O2, antibiotics, hydration, avoiding over hydration, albuterol nebulisers SOS, and simple transfusion
Acute neurological events
Out of the SCD diagnosed patients, 10-14% suffer stroke. The most common age group is between 5-10 years. However, the first episode can occur anytime upto 20 years. Refer patient for urgent exchange transfusion. Children with a history of stroke need long term transfusions, once every 3-4 weeks, to maintain Hb S concentration below 30 %, to reduce the risk of recurrence.
Fever
In SCD patients fever must be taken seriously as death from sepsis is common. Admit the patient (child or adult) if temperature is > 100 F, and treat as follows:
- Immediate evaluation with history and physical examination, CBC and, if available, blood culture, and urine culture when urinary tract infection is suspected
- Promptly administer IVceftrioxone + cloxacillin +/- gentamycin and oral azithromycin as Gram + including Staph aureus and Gram – pathogens need to be covered. Consider antimalarials in malaria endemic areas SCD patients have normal T cell and B cell function, so the risk of acute infection is generally limited to the micro-organisms mentioned above. Opportunistic infections are infrequent.
Paediatric care
SCD being a genetic disorder affects individuals from the moment they are born. It is important to start interventions at an early age in order to improve outcomes as well as life expectancy. The following section gives an overview of the course of the disease as well as interventions to be considered as children affected by SCD grow.
Children Under 5
- Ideally children should be identified through newborn screening. In the absence of such programs, children should be tested as early as possible.
- After testing, results should be properly communicated to parents,
- The site and nature of pain changes with the age of a child. Children under the age of 3 will frequently cry. Look out for swelling of feet and fingers and for pain.
- Teach caregivers to check spleen size and ask them to check daily for any increase/ change in size.
- Immunisation schedule should be up to date and, if available, pneumococcal conjugate vaccine can be offered, ideally from birth to 2 years of age. Serotypes specific to India are also now available. After 2 years of age, the pneumococcal polysaccharide vaccine should be given.
- If stroke is common in your area, transcranial doppler (TCD) scans are advised starting at 2 years of age and continuing up to late teens. Inexpensive, battery operated (non- imaging TCD) machines are available now-a-days that can be used.
- Children are also started on penicillin prophylaxis to prevent serious infections as soon as possible after diagnosis.
- Dosage for Penicillin - Pentids is given as follows:
- 65 mg twice a day ill 12 months
- 125 mg twice a day for children under 2 years
- 250 mg every twice a day between 2 to 5 years or Amoxicillin 20 mg/kg/day
- Penicillin should be continued lifelong if a patient has had a splenectomy
- Alternatively, if a patient is allergic to penicillin azithromycin 10 mg/kg/day
Children between 5 to 10 years
- Site of pain often shifts to sites of large joints like elbows, ankles, knees and wrists.
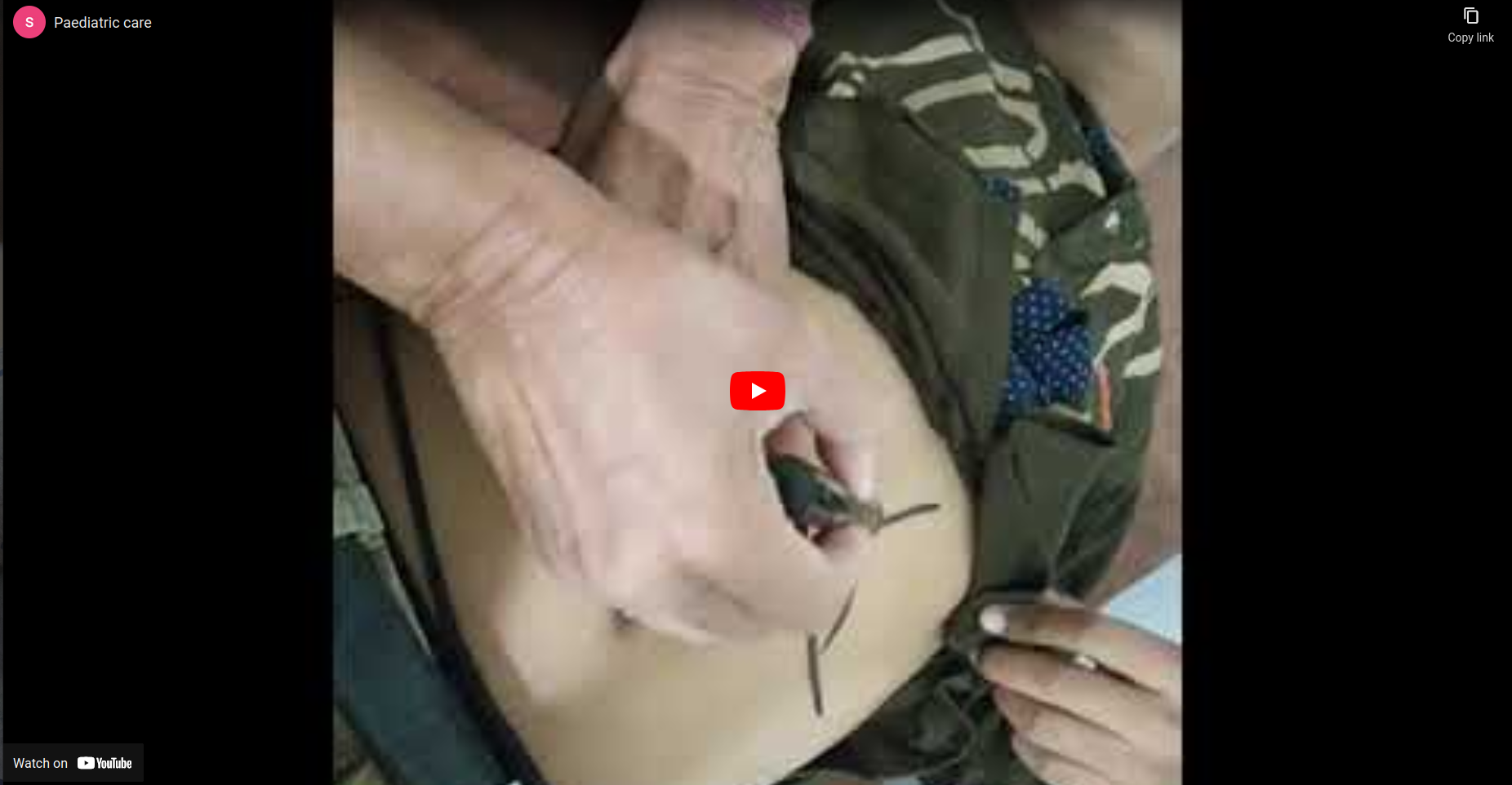
- There is a high risk of sequestration of organs, especially of the spleen and liver. First, the spleen increases in size (see diagram and watch the video), the patient starts looking pale and then goes into shock in a few hours
- Patient needs to be taken to the nearest health facility immediately for blood transfusion. If blood is unavailable start IV Normal saline and refer to hospital with a Blood bank
- Avoid over-transfusion. The blood in the spleen goes back into the circulation usually in 24-48 hours.
- If the child has had 2 sequestration events over a period of 1 year, refer for 10 splenectomy
- Anaemia is common in this period and could be caused by hemolysis, nutritional deficit, sequestration, jaundice, and infection such as malaria, parvovirus etc. Find the cause and treat it, avoiding unnecessary transfusions
- Stop penicillin at age 5 unless patient has had splenectomy when it is continued for life
- The following routine tests are recommended:
- Annual urine microalbumin after 5 years of age.
- pulmonary function (spirometer)
- cognitive assessment.
- As a proxy to monitor microinfarcts of the brain, evaluate cognitive performance:
- With reasoning and memory tests
- Noting school performance, such as time taken to complete assignments, and trajectory of grades on exams and monitoring time taken to learn new skills
- Flu and typhoid, if endemic in your region, the vaccines should be given annually, if available.
Children from 10 to 18 years
- In addition to long bones and joints, the site of pain might shift to vertebra and chest areas.
- Any pain in the hips should be taken seriously, this could be an early indication of avascular necrosis.
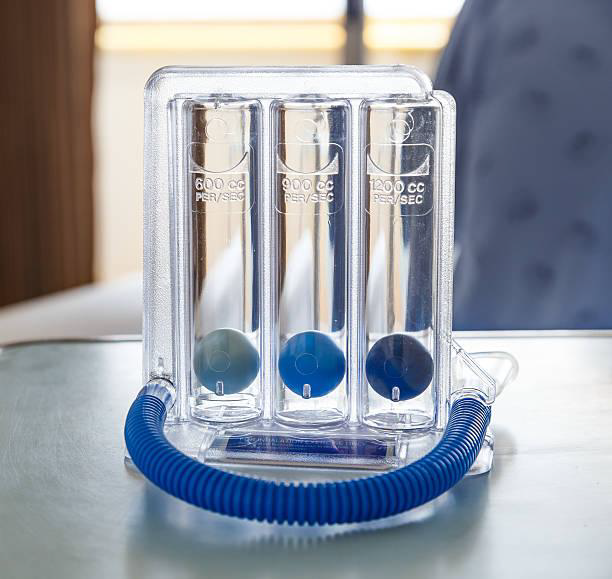
- Acute chest syndrome is quite common at this age in South Asia. Advise patients to use a spirometer every day starting at the age of 5.
- Flu and typhoid, if endemic in your region, the vaccines should be given annually, if available.
During adolescence, puberty can be delayed. Patients may experience physiological and sexual development differently than their peers, for example the onset of menarche for girls occurs later. Patients may need additional psychological support and care.
Image 2: Digital Thermometer, 2b: Spirometer
Adolescence and transitioning to adult care
Adolescence is a very difficult period for SCD patients, over and above the usual issues faced by young people at this stage. They want to be like their peers and be ‘normal’ and do everything their friends, siblings, cousins do. Many children may drop out of school, some show signs of depression, and others might start abusing substances (depending on local practices in their community).
Patients may also stop complying with medical advice and stop taking their medications regularly. A majority of patient are anxious about delays in physical growth and signs of puberty, including growth of body hair (boys) and menarche (girls). They may not be able to discuss these issues with their parents. This can lead to emotional anxiety and even distress.
Often parents have similar anxieties but discuss the matter with healthcare practitioners without involving the young person. Healthcare practitioners have their own attitudes and biases about what the future of these young people might be like and can give inappropriate advice. Patients need to be reassured that their physical growth and sexual maturity might be delayed but it will occur and there are medications available if need be.
A lot of emotional, psychological and peer support is required for patients as well as their parents/family during this phase. Appropriate, information, counselling and a referral to an endocrinologist, if need be, should be considered. For students who are unable to keep up with school due to poor health, options for vocational training need to be explored.
Hydroxyurea (HU) Recommendations
5.1. It is important to educate all patients with SCD and their family members about the benefits, importance of compliance, dosage and laboratory follow up of HU. Sexually active adult male and females should be counselled regarding the need for contraception when on hydroxyurea treatment.
Hydroxyurea should be avoided in
- Pregnancy or sexually active patients unwilling to use contraception.
- Active liver disease (HBV or HCV infection).
- Toxicity or hypersensitivity
- History of non-compliance with medical care.
5.2. It is important to have an established prescribing (dosage and how to take it) and monitoring protocol for Hydroxyurea therapy in SCD patients. SCD is a highly variable condition, the indications for starting hydroxurea at any age after 6 months are:
- One admission with VOC, anemia, ACS, stroke, or AVN recurrent sickle cell-related pain that interferes with daily activities of life
5.3. Baseline HU investigations before starting HU
- Hb electrophoresis and quantitative HbF % if available
- CBC, ALT, serum creatinine, microalbuminuria
5.4. Starting Hydroxyurea
This maximal therapeutic dose (MTD) is a dose at which hydroxyurea leads to the greatest benefit without toxicities or side effects and where there is acceptable degree of marrow suppression (decrease in WBC and platelet count).
Based on data from Indian studies (Jain et al., 2013), the recommendations are to start the patients on 10 to 15 mg/kg/day of HU as a single morning dose.
If there is still pain (less than 25% improvement, clinically as well as subjectively) dose is increased by 5 mg/kg every 12 weeks till maximum dose of 30mg/kg is reached or until evidence of hematological toxicity (see below)
In those is situations where a patient on hydroxyurea therapy is not likely to follow up or cannot be monitored, the patient should be given a fixed low dose hydroxyurea therapy (10 mg/ kg) without escalation.
HU dosage for paediatric patients
In India hydroxyurea comes as a 500 mg capsule. Follow these steps in order to prepare a lower dose of the medication for paediatric patients:
- Take a 500 mg capsule, open it, and place the powder on a clean sheet of paper.
- Divide it into the daily dose as prescribed by weighing it on a digital scale
Wrap each daily dose separately and give the patient one month’s supply. The video demonstrates how to make a paediatric dose.
5.6. Essential Monitoring while on Hydroxyurea
- All the patients started on HU should be monitored with CBC within 3 weeks of starting treatment, at least once every three months thereafter and every time the dose is increased.
- SGPT and serum creatinine should be checked every six months. The aim is to maintain an absolute neutrophil count (ANC) above 2000/ml or a WBC above 3500/ml and platelet count above or at 80,000/µL. If WBC/platelet count drops below the above limits, HU is withheld and restarted at 5mg lower than the previous dose when WBC recovers to 2500/ml or higher, and platelet count above 80,000/ml SGPT, serum creatinine every six months.
Response to Hydroxyurea
- Clinical response with HU treatment will take 3 to 6 months. So dose should be increased after a minimum of 3 months
- A 6-month trial on the maximum tolerated dose (MTD) is necessary before considering drug discontinuation.
- Increase in MCV can be used as a proxy for compliance.
- Hydroxyurea should be continued during hospitalisation unless the patient has toxicity.
It is important to note that a majority of the cases of non-response are due to non-compliance to therapy or a failure to escalate to maximum tolerated dose (MTD).
In addition to hydroxyurea, the following are often prescribed as part of adult care:
- Folic acid 5mg/day
- Iron tablet (Ferrous sulfate) 200mg/day only if the patient is iron deficient as demonstrated by hypochromia and microcytosis of RBCs on peripheral smear
- For pain Paracetamol 500mg can be taken up to three times a day as needed.
- Adult vaccinations records should also be kept and anyone above the age of 19 who has not yet received pneumococcal vaccine should be given one.
Recommended pneumococcal vaccine schedule
- First dose of pneumococcal conjugated vaccine (PCV13) given by intramuscular route. After a minimum gap of 8 weeks polysaccharide pneumococcal vaccine PPSV23 is given by intramuscular route.
- Second dose of PPSV23 can be given by intramuscular route five years after the first polysaccharide pneumococcal vaccine.
Adult Laboratory information
- Baseline haemoglobin levels of each patient should be documented at a steady state (well) as this is important for the long term management and setting a norm for the patient. Expecting patients to maintain an haemoglobin levels of of 10-11 can be unrealistic and result in unnecessary and harmful transfusions (see section on transfusions below)
- Hb electrophoresis and quantitative HbF % if available
- CBC, ALT, serum creatinine, microalbuminuria
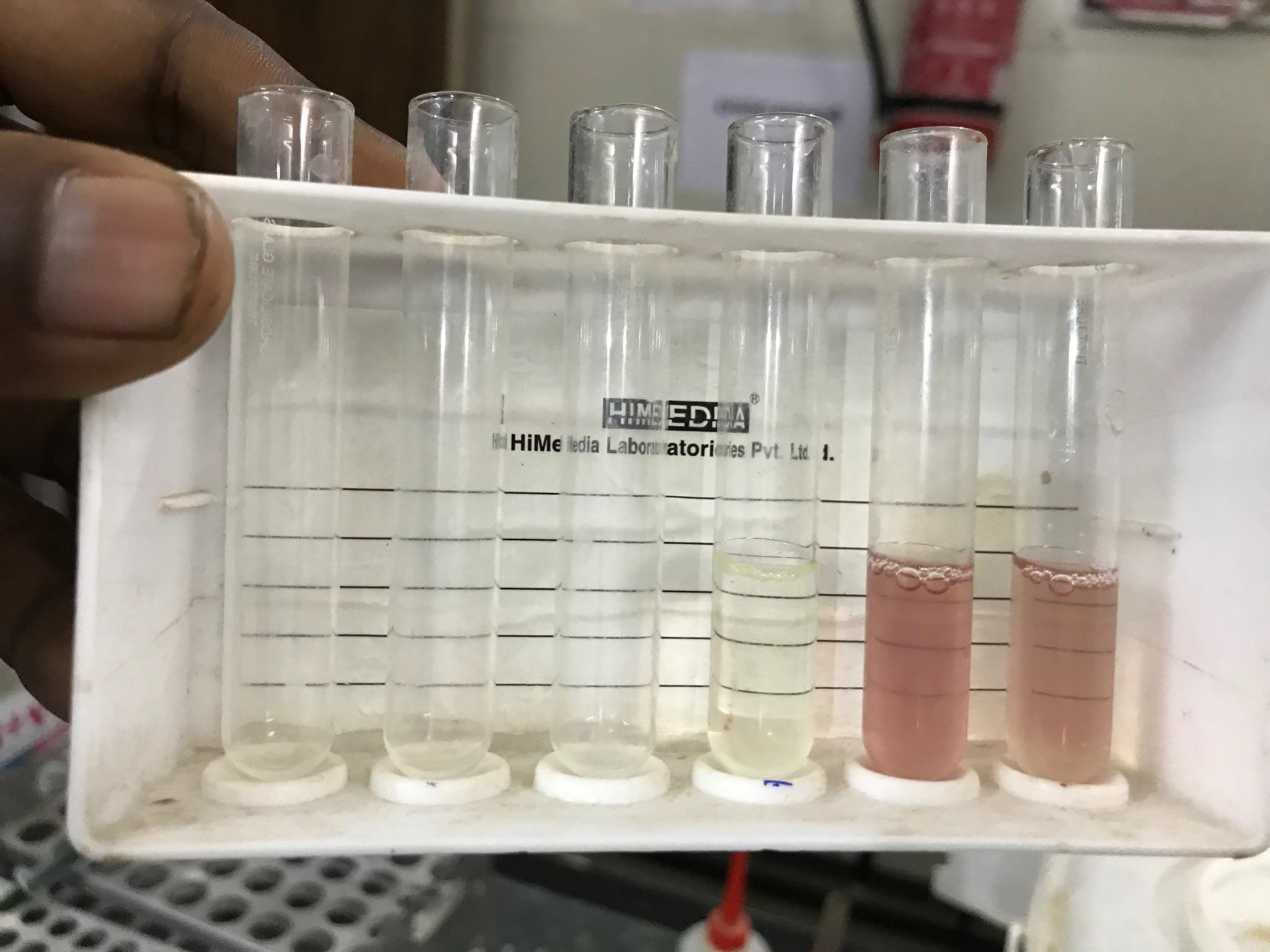
Image 3: Solubility test
Pregnancy Care (pre, antenatal and postnatal)
SCD is a variable condition and it is important for healthcare practitioners to be supportive of women who are or want to be pregnant. Some patients might even be under family pressure to become pregnant soon after marriage. The important point is to provide information and supportive care, before, during and following birth. Training community health care workers and those associated with maternal care in this area is crucial
During pregnancy there is an increased risk of
- Acute painful crises
- Premature labour
- Spontaneous miscarriage
- Common Infections in the area
- Thromboembolic events
- Antepartum and postpartum haemorrhage
- Pre-eclampsia and
- Pregnancy-induced hypertension
- Maternal mortality as a result of the increased risks mentioned above .
There is also an increased incidence of perinatal mortality and intrauterine growth retardation of the fetus.
Antenatal care
- Hydroxyurea should be stopped at least 3 months before conception and restarted after the cessation of breastfeeding. It can however be restarted during breastfeeding if the mother has an increased incidence of crisis.
- Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers should be stopped before conception. If already pregnant, switch to another anti-hypertensive.
- Regular antenatal checkups with all the routine antenatal tests are required blood pressure and urinalysis should be performed at each visit
- Persistent vomiting can lead to dehydration and crisis. If severe and causing ketoisis, IV fluids should be given.
- ron supplementation should only be given if there is laboratory evidence of iron deficiency and no history of frequent blood transfusions.
- Folic acid 5mg once per day should be continued
- Low-dose aspirin 75mg once daily starting at the 12th week of gestation can be prescribed to reduce the risk of developing pre-eclampsia.
- Non-steroidal anti-inflammatory drugs (NSAIDs) should be prescribed only between 12 and 28 weeks of gestation owing to concerns regarding adverse effects on foetal development.
- Blood transfusion in pregnancy is indicated only if there is a drop in haemoglobin of over 2gm from the patient’s baseline (refer to section on blood transfusion below).
- Perform ultra-sound examinations when possible
- Offer a viability scan between 7 to 9 weeks of gestation.
- Offer routine first-trimester scan 11 to 14 weeks of gestation and a detailed anomaly scan at 20 weeks of gestation
Type of delivery and timing
Should have pre-anesthesia evaluation in the third trimester of pregnancy to assess the risks of possible C-section
Vaginal delivery is possible for those with SCD
- Vaginal delivery is viable for pregnant women with SCD with a normal growing foetus after 38 weeks of gestation.
- Ideally delivery should take place in a centre familiar to the patient where an obstetrician, blood bank, and facilities for C-section and paediatrician are available.
- Blood should be cross matched for delivery. At least 4 units should be available in the event of post -partum haemorrhage.
- If Caesarean section is needed spinal or epidural anaesthesia is recommended
Avoid the use of pethidine as it is associated with an increase in seizures. Tramadol and morphine are safe to use.
Patient’s requests for specific pain medicines or doses are most commonly due to past experience and not inappropriate drug-seeking behaviour. Pain scales can be useful for assessment.
Build trust by believing the patient is in pain and show them you are there to help. Empathetic body language is essential (eye contact, facial expression, gestures) Be patient when asking questions; it is often difficult to speak when in severe pain. Pain can make anyone irritable, impatient, or upset.

Image 4: NHM Sickle Cell Management program (2017-18), kurkheda, Gadchiroli
Blood Transfusion
Blood transfusion remains an important therapeutic intervention for patients. Emergency simple transfusion is indicated for the management of acute anaemia and acute chest syndrome. Acute anaemia results from splenic sequestration where the spleen suddenly enlarges, from temporary bone marrow failure or from sudden hemolysis.
Defining baseline haemoglobin
- Transfusion in acute anaemia should be given if the Hb drops 2gm% or more from the usual or baseline Hb. Blood should only be given to bring the Hb back to the baseline. This is very often around 8gm% in most SCD patients. There is no need to transfuse such a patient to 10gm Hb.
- If the lab does not do reticulocyte count then it will not be possible to identify bone marrow failure. The patient should be admitted and Hb checked daily and if there is no further drop, the patient can be discharged.
- Transfusion is not indicated for the management of an uncomplicated pain crisis. Frequent transfusions lead to iron overload and liver failure, alloimmunization (formation of antibodies against non–self-antigens on donor RBCs), and vaso-occlusive episodes and life-threatening hemolysis.
Evaluation and management of iron overload
Those requiring frequent transfusions (including patients who are co-heterozygous for sickle cell and beta-thalassemia) must be monitored regularly for iron overload and assessed for benefits of chelation therapy. A patient, who receives 10 units of pRBC units per year, receives an excess of 2 grams of elemental iron that can damage heart, liver and other vital organs in the long term.
Serum ferritin reflects the overall iron stores in the body tissues and thus is a useful indicator of iron storage. Serum ferritin is an acute phase reactant, so its value varies with the presence of any infection or inflammation in the body. Hence a single value of this test is not useful. The trend should be monitored by performing a ferritin test once in six months for those receiving multiple transfusions.
- Detecting iron overload in organs such as the liver and heart requires. Referral may be necessary for MRI and possibly liver biopsy.
Iron Chelation
The serum ferritin levels should be assessed after 10 to 15 transfusions and chelation therapy should be initiated when the serum ferritin value is more than 1000μg/L. Serum ferritin monitoring is mandatory with all chelation therapy.
Blood Transfusion for Acute and Chronic Complications in SCD
| Complication | How to transfuse |
| Acute Chest Syndrome: Symptomatic, with decreased Hb of 1g/dL below baseline | Simple |
| Anemia, Symptomatic | Simple |
| Aplastic Crisis | Simple |
| Spelnic Sequestration + severe anemia | Simple |
| Acute Chest Syndrome: Symptomatic, Severe (Oxygen Saturation less than 90% despite supplement Oxygen) | Exchange |
| Hepatic Sequestration | Exchange or Simple |
| Intra-hepatic cholestasis | Exchange or Simple |
| Multi-system Organ Failure | Exchange or Simple |
| Stroke, Acute | Simple or Exchange |
| Chronic : Stroke risk | Chronic program, Exchange or Simple |
Source: ASH, Expert panel Report 2014
Genetic Counselling
Genetic counselling aims at providing information and support to individuals and families about genetic risks of passing on the gene/ disorder to the children. Counselling couples at risk is complex because SCD can vary from being relatively severe to mild/asymptomatic in presentation. This must be clearly explained to the parents.
Counselling should be offered :
- After the diagnosis of trait or disease
- Before pregnancy or as early in pregnancy as possible (see the section on antenatal care).
- During antenatal testing when both mother and father are known/diagnosed as sickle cell carriers
- Before and following prenatal diagnosis to inform the couple about the diagnosis of the fetus (see the section on prenatal diagnosis)
For newborn screening
Counsellors should know the difference between SCD and trait. Carriers (traits) should be reassured that they do not have/will not have any of the problems of SCD. They need to know the risks of marrying a person with a trait.
For SCD patients:
- Explain the clinical presentation, variability, severity, and consequences of the disorder.
- the autosomal recessive inheritance pattern.
- the risks and meaning of the carrier state
- Providing emotional and social support to reduce the family’s anxiety
Counsellors and health practitioners should be able to answer basic questions, in a simple, language:
- Why is our child suffering from the disease when both of us normal?
- Is there a cure for the disease?
- What is the cost involved for treatment?
- What treatment options are available, are these lifelong?
- Are these treatments available near our home?
- Can the next child also have a similar disease?
- Is there any way to prevent it?
Inheritance of SCD
Sickle Cell Disease is an inherited blood disorder. Hence, a child is born with it. It is not transmitted from one person to another through any other source. It is a recessive gene disorder. A baby inherits two copies of the gene (one from each parent) to have the disorder or one copy to have a trait / be a carrier. The parents can have a simple blood test, to easily find out if there is a risk of their children having the disease. The risk in each pregnancy is important to understand in order to explain the risk in subsequent pregnancies Prospective parents can face the following scenarios, depending on their own haemoglobin status.
- AA – both parents have normal haemoglobin - AA - there is absolutely nothing to worry since the children will be fine
- AS – if one parent is AS / has a sickle cell trait, then there is a 25% chance that a child might also be a carrier
- If both parents are AS - then there is a 25% risk that a child might have SS, 25% chance that it will be AA, and a 50 % that it might have a trait (AS).
- SS – IF both parents have sickle cell disease (SS) then all children have the same, 100% risk of having the SS.
- If one parent is SS and the other is AS, a child will have a 50% risk of inheriting a trait (AS) and 50% risk of having SS
Since the disease is transmitted to the children only from their parents, genetic counselling becomes extremely important in the management of sickle cell disease. (More details about counselling are given in the next section).
Ethical and Religious Issues:
Genetic counselling should be “non- directive”, where the counsellor presents the information regarding patterns of inheritance and allows the individual or the couple to make their own decisions on marriage and procreation. How they interpret the genetic information and risk is guided by social, cultural, and religious factors as well as the influence of elders in the family.
Informed Consent:
The health practitioner/ counsellor must seek informed consent before any tests (including screening) or procedure is undertaken. Consent can be written, verbal, or non-verbal.
Reproductive Counselling
Health practitioners need to be sensitive to the personal and social circumstances and aspirations of the patients. Often women, across the rural/urban divide, can be under social pressure to have children soon after marriage, and delays in childbearing can affect their position in the family. It might be useful to initiate a discussion about what the woman and her husband want, and how to achieve these goals.
Given the increased risk of adverse pregnancy outcomes in SCD and the risk of maternal morbidity and mortality, several recommendations are relevant for women with SCD and their partners. It is important to be supportive in case the woman wants to have a baby rather than discourage her or overtly/ covertly persuade her not to have children or terminate a pregnancy without giving her the choice of having a prenatal test.
Premarital testing
At times, individuals might have been screened in the community and know their carrier status and want their prospective partner to be tested. There might be sickle cell in the family, resulting in premarital tests. Screening results should not be shared with anyone except the patient, as public disclosure can have serious repercussions for the person. Carrier couples should know the risks outlining the scenarios presented above (1. One partner is a carrier 2. Both partners are carriers 3. One is a carrier and the other has SCD, 4. Both have SCD)
Contraception
In women with SCD, regular use of contraception is advised to reduce the health risks associated with an unintended pregnancy. Hormonal contraceptives may also decrease menstrual blood flow, leading to higher haemoglobin levels. The use of progestin-only
hormonal contraceptives lowers the risk of thromboembolism compared to the use of oestrogen-containing contraceptives and has been shown to be safe for women with SCD. However, if the benefits are considered to outweigh the risks, combined hormonal contraceptives (pills, patches, and rings) may be used in women with SCD. A history of stroke is a contraindication to combined hormonal contraception,
Intrauterine devices (IUDs) and intrauterine implants carry modest risks associated with the insertion procedure, while sterilization carries risks associated with the surgical procedure. There is no evidence that IUDs pose an increased risk for women with SCD.
Explaining the risks during pregnancy
- Pregnancy in SCD is considered high risk for the mother and the baby.
- For the mother, these risks are increased frequency of pain crises, thrombosis, infections, preeclampsia, and death as compared to women who do not have SCD.
- Possible adverse outcomes include foetal (intrauterine) growth restriction, preterm delivery, and stillbirth.
- Additional foetal surveillance is required during pregnancy
Social and emotional aspects of SCD: How to support patients and family carers
SCD affects children, adults and their families differently, depending on the severity of illness, their financial situation, medical and social support.
- Health practitioners often tell parents that children with SCD do not survive beyond their late teens- early 20s. Describing SCD as a dreadful disease and a burden on the family, may result in poor expectations of health and the future of the child
- Likewise, negative health promotion posters depicting debilitating images of SCD as a burden on the family and society reinforce these collective attitudes and stigma.
- Families often treat their children with SCD differently, expecting their lives to be brief. Since these children are more likely to miss school or exams, their education and self esteem suffer.
- It is therefore important to challenge these negative images and provide parents with positive examples of children who have faced these challenges and succeeded in school.
- School teachers need to be informed and supportive. They should counsel regarding appropriate vocational training and employment opportunities
- Health practitioners need to be aware of The RPWD ACT and the right to education, employment, and the process of getting a disability certificate and pension.
- It is important to remember that SCD is a variable condition and whist a disability certificate for life sounds like a good option, the level of disability might change in the future. Hene, there is merit in advocating the option of a review.

Image 5: ASHWINI Sickle Cell Centre
Acknowledgements
We would like to thank members of our advisory board – Karl Atkin, Johnny Oomen, Preeti Shinde, Saraswathi C.D, Sunil Ghanmode, Kareem Karassery, Yujin Kushmi, Pankaj Kavikumar and Sanghamitra Das - for their time, invaluable support and insightful comments and suggestions on the content and successful dissemination of the toolkits. Smitha M, Priyankar C, Pushpanathan J, Ajith JS and Sunu G deserve special thanks for bringing the event together so seamlessly across India, York, Baltimore and Nepal, providing superb support with IT, multi-lingual translations and field visits. Others have contributed in many ways, sharing their experiences and being part of the conversations culminating in the idea for this workshop over the years. Finally, we thank Grace Cooper, our brilliant student intern at the University of York, for her inputs and excellent formatting of both the documents.
References
ASH, Expert panel Report 2014. [Online]. Available at: https://www.nhlbi.nih.gov/health-topics/evidence-based-management-sickle-cell-disease
Erhan E, Inal MT, Aydinok Y, Balkan C, Yegul I. (2007) Tramadol infusion for the pain management in sickle cell disease: A case report. PaediatrAnaesth. 17(1). 84–6.) [Online]. Available at: 10.1111/j.1460-9592.2006.02030.x
Government of India. 2016a Prevention and Control of Haemoglobinopathies in India: Thalassaemias, Sickle Cell Disease and other Variant Haemoglobins (National Health Mission Guidelines on Hemoglobinopathies in India). New Delhi: Ministry of Health & Family Welfare. http://nhm.gov.in/images/pdf/programmes/RBSK/Resource_Documents/Guidelines_on_Hemog lobinopathies
Jain DL, Apte M, Colah R, Sarathi V, Desai S, Gokhale A, Bhandarwar A, Jain HL, Ghosh K. (2013) Efficacy of fixed low dose hydroxyurea in Indian children with sickle cell anemia: a single centre experience. Indian Pediatr. 50(10):929-33. [Online]. Available at: 10.1007/s13312-013-0264-0.
Joseph F Standing, Imogen Savage, Deborah Pritchard, and Marina Waddington. (2015). Diclofenac for acute pain in children. (Cochrane Database Syst Rev).CD005538. PMCID: PMC6564078, PMID: 26134060. [Online]. Available at: 10.1002/14651858.CD005538.pub3
Raman V, et al. (2021). BMJ Global Health. 6:e004322. [Online]. Available at: 10.1136/bmjgh-2020-004322